- Angelika Batzner, Hans-Joachim Schäfers, Konstantin V. Borisov, Hubert Seggewiß: Hypertrophic obstructive cardiomyopathy—the role of myectomy and percutaneous septal ablation in drug-refractory disease. In: Deutsches Aerzteblatt Online.
- Authors/Task Force members, PM Elliott, A Anastasakis, MA Borger, M Borggrefe, F Cecchi, P Charron, AA Hagege, A Lafont, G Limongelli, H Mahrholdt, WJ McKenna, J Mogensen, P Nihoyannopoulos, S Nistri, PG Pieper, B Pieske, C Rapezzi, FH Rutten, C Tillmanns, H Watkins: 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). In: European Heart Journal. Band 35, Nr. 39, 2014, S. 2733–2779
- American College of Cardiology / European Society of Cardiology Clinical Expert: Consensus Document on Hypertrophic Cardiomyopathy. In: Journal of the American College of Cardiology. Band 42, Nr. 9, 2003.
- F. Sedaghat-Hamedani, E. Kayvanpour, O. F. Tugrul, A. Lai, A. Amr, J. Haas, T. Proctor, P. Ehlermann, K. Jensen, H. A. Katus, B. Meder: Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. In: Clinical Research in Cardiology. Juli.
- C. Hengstenberg: Genetik der familiären hypertrophischen Kardiomyopathie. (PDF; 216 kB) In: Dtsch. Arztebl. Band 93, Nr. 9, 1996, S. A-532 / B-430 / C-406.
- H. Kuhn, J. Mercier, E. Köhler, H. Frenzel, W. Hort, Franz Loogen: Differential diagnosis of hypertrophic cardiomyopathies: typical (subaortic) hypertrophic obstructive cardiomyopathy, atypical (midventricular) hypertrophic obstructive cardiomyopathy and hypertrophic non obstructive cardiomyopathy. In: Eur Heart J, Band 4, Suppl F, 1983, S. 93–104.
- Michael Schäfers et al.: Nichtinvasive kardiale Bildgebung. In: ecomed Medizin. 2008, ISBN 978-3-609-16282-9.
- Iacopo Olivotto, Artur Oreziak, Roberto Barriales-Villa, Theodore P. Abraham, Ahmad Masri, Pablo Garcia-Pavia et al.: Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. In: The Lancet. Band 396, Nr. 10253, 2020, S. 759–769, doi:10.1016/S0140-6736(20)31792-X.
- Christian Mewis, Reimer Riessen, Ioakim Spyridopoulos (Hrsg.): Kardiologie compact – Alles für Station und Facharztprüfung. 2. Auflage. Thieme, Stuttgart/ New York 2006, ISBN 3-13-130742-0, S. 396–397
- L. Faber, Hubert Seggewiss, F. H. Gietzen, H. Kuhn, P. Boekstegers, L. Neuhaus, L. Seipel, Dieter Horstkotte: Catheter-based septal ablation for symptomatic hypertrophic obstructive cardiomyopathy: follow-up results of the TASH-registry of the German Cardiac Society. In: Zeitschrift für Kardiologie Band 93, Nr. 1, Januar 2004, S. 23–31;
- U. Sigwart: Non-surgical myocardial reduction for hypertrophic obstructive cardiomyopathy. In: The Lancet. Band 346, Nr. 8969, 22. Juli 1995, S. 211–214.
- H. Kuhn: Transcoronary ablation of septal hypertrophy (TASH): a 5-year experience. In: Z. Kardiol. Band 89, Nr. 6, Juni 2000, S. 559–564.
- A. Oto, K. Aytemir, A. Deniz: New approach to septal ablation: glue (cyanoacrylate) septal ablation. In: Catheter Cardiovasc Interv. Band 69, Nr. 7, 1. Juni 2007, S. 1021–1025.
- T. Lawrenz, H. Kuhn: Endocardial radiofrequency ablation of septal hypertrophy. A new catheter-based modality of gradient reduction in hypertrophic obstructive cardiomyopathy. In: Zeitschrift Für Kardiologie. Band 93, Nr. 6, Juni 2004, S. 493–499
- M. Emmel, N. Sreeram, J. V. deGiovanni, K. Brockmeier: Radiofrequency catheter septal ablation for hypertrophic obstructive cardiomyopathy in childhood. In: Zeitschrift Für Kardiologie. Band 94, Nr. 10, Oktober 2005, S. 699–703
- Claudia Strunk-Mueller, Frank H. Gietzen, Horst Kuhn: Schrittmachertherapie der hypertrophisch obstruktiven Kardiomyopathie. In: Herzschrittmachertherapie und Elektrophysiologie. Band 15, Nr. 1, Juli 2004, ISSN 0938-7412, S. i47–i53
- Bernard J. Gersh, Barry J. Maron, Robert O. Bonow, Joseph A. Dearani, Michael A. Fifer, Mark S. Link, Srihari S. Naidu, Rick A. Nishimura, Steve R. Ommen, Harry Rakowski, Christine E. Seidman, Jeffrey A. Towbin, James E. Udelson, Clyde W. Yancy: 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy. In: American College of Cardiology / American Heart Association (Hrsg.): Circulation. Band 124, Nr. 24, 13
Hypertrophe Kardiomyopathie: Was ist das und wie äußert sie sich? + Risiken
Fotoquelle: Getty images
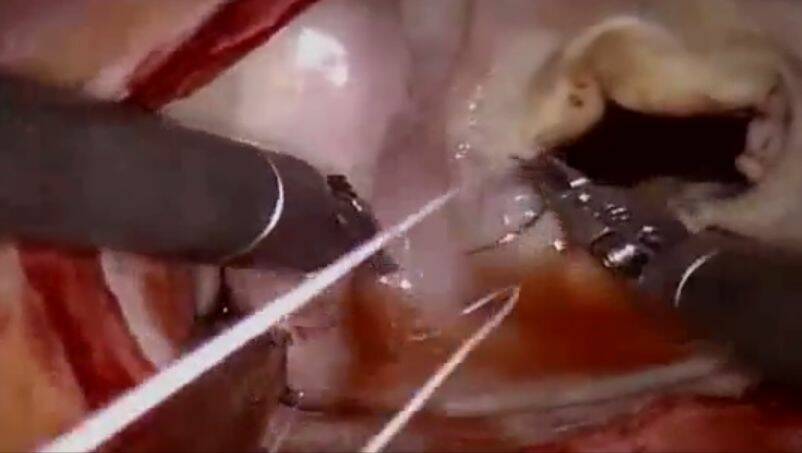
Behandlung der hypertrophen Kardiomyopathie
Mehr anzeigenHypertrophe Kardiomyopathie wird behandelt von oder durch
Andere Namen
hypertrophische Kardiomyopathie; HCM